我们日常接触到的客户发布整改,有些项目问题点少,但是非常非常的难改,有些看上去几十个上百个问题点,但是整改起来只是耗费时间,并无无法攻克的硬骨头。所以从来都不是问题越少越好改,问题越多越难改。
很多人问黄老师,我们在做技术文件时候,怎么才能避免出现无法挽救的硬伤呢?我认为核心的问题点在于:我们是否能够建立并维护从用户需求规范到功能需求规范、风险分析、临床评估和通用安全与性能要求之间的双向可追溯性。而这是我们向监管机构证明证据链的完整性和一致性的唯一方法。
那么怎么才能做到这一点呢,黄老师曾经参加BSI的一个培训,他们给了一个图表:
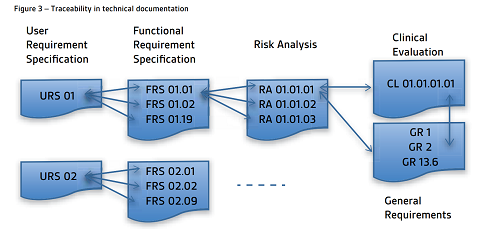
这个表当时老师一笔带过,没有细说,也很少有客户真正的用到了实际的工作中。而使用这一方式,已经成为欧杰老师的标准作业方式。我们以一个心率监控仪的实际案例来说明它:
1:URS-001:用户需要设备能连续监测心率。
FRS-001.01:设备必须能够每秒钟测量并记录一次心率值。
RISK-005.01:心率测量不准确导致误报的风险。
VER-012.01:验证心率测量精度的测试。
GSPR-9.1:适用于诊断功能的器械,其准确度必须得到验证。
在识别了上述需求后,我们尝试着建立一个可追溯性矩阵
|
用户需求
|
功能需求
|
相关风险
|
验证与确认
|
临床评估
|
适用的GSPR
|
|
URS-001:连续监测心率
|
FRS-001.01:
每秒测量心率
|
RISK-005.01:测量不准确
|
VER-012.01:精度测试报告
VAL-020.01:临床性能研究
|
CL-003.01:评估心率数据的临床有效性
|
GSPR-9.1,
GSPR-10.1
|
|
URS-002:设备使用安全
|
FRS-002.01:电气隔离
|
RISK-010.01:电击风险
|
VER-015.01:电气安全测试
|
CL-005.01:评估使用安全性
|
GSPR-10.4
|
|
URS-003:材料生物相容
|
(不适用)
|
RISK-015.01:过敏反应
|
VER-025.01:生物相容性测试报告
|
CL-004.01:评估生物相容性数据
|
GSPR-10.4
|
这个矩阵如何实现“双向”追溯?
1:正向追溯:从左到右。可以清晰地看到一个用户需求(URS-001)是如何通过功能需求(FRS-001.01)被实现,其相关风险(RISK-005.01)如何被控制和验证(VER-012.01),并最终在临床评估(CL-003.01)中证明其安全有效性,同时满足了哪些GSPR。
2:反向追溯:从右到左。如果监管机构问“你们是如何满足GSPR-9.1的?”,你可以通过矩阵快速定位到所有相关的验证测试、风险控制措施、功能需求和最初的用户需求,形成一个完整的证据链。
如何将风险管理作为核心枢纽?
风险管理系统是连接设计输入(URS/FRS)和输出验证(临床前/临床)的桥梁,黄老师这里特别要提醒大家下面两个问题点:
1:每个功能需求都应考虑其可能引发的危害。
2:每个风险控制措施(如在软件中加入报警功能)本身可能成为一个新的设计输入或验证要求,需要被追溯。
如何将临床评估与设计和风险紧密关联
黄老师在很多企业培训中,都会反复强调:临床评估不应是一个独立的报告,而应是整个证据链的顶峰。
1:临床评估计划应明确需要解决哪些由风险分析提出的问题。
2:临床评估报告应引用设计和验证阶段的数据(如性能测试),并证明风险已被控制在可接受水平,受益大于风险。
做医疗器械注册,无论是国内的NMPA,还是欧盟的MDR,还是美国的FDA, 最忙碌的永远不应该只是法规部门,也应该包含设计部门,只有把技术文件的形成无缝地整合到质量管理体系和设计控制流程中,并确保做到:
1:在设计开发流程中,明确要求在每个阶段(如设计评审、设计转换)检查可追溯性矩阵的完整性。
2:任何设计变更都必须经过评估,并更新可追溯性矩阵,以确保整个证据链的持续一致性。
那么,技术文件不合格出现再多的问题也都是细枝末节的小问题,一般都不会出现无法进行下去的大问题。
如果大家对这个话题感兴趣,欢迎大家留言区和我们一起探讨。









